Hello!
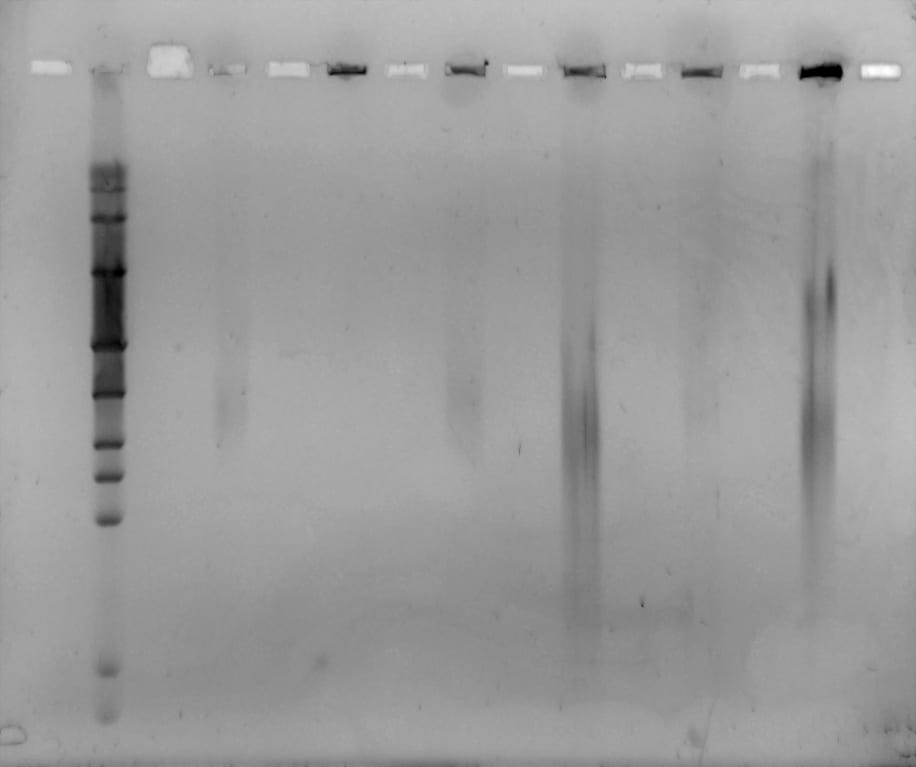
Today was our last day in the lab! 🙁 We filmed our latest and final videoblog, removed the gel, stained and viewed it. We saw some separation from all but one of the plugs, however separation was very blurred. We decided to run the same samples again, but for longer and with a higher switch interval. We set up and ran a new gel for 18 hours, at 6V/cm, with a 1-30 second switch interval, a 1.4% gel with modified buffer flow.
Mike will remove the gel tomorrow, stain it and view for us. Hopefully we will see better separation, from the samples containing enough DNA.
The last 10 weeks have been amazing – i have learnt so much since i started and it’s going to be really odd to not be in the lab everyday. I have really enjoyed working through a project, having to work on problems and figure out solutions. Then getting to see the good and bad results. I am very impressed with myself, as i set out to blog everyday i was in the lab with what i have done, the good and the bad! and i have actually managed it! 🙂 I wasn’t sure about whether i would get on with videoblogging at the beginning, but i have really enjoyed filming them every week – although most the content was never included as we ALWAYS got distracted! I really hope those who have watched them have found them as interesting and enjoyable as we did.
It has been a very rewarding experience, and i am so grateful to everyone who has made the last 10 weeks, the best summer – you all know who you are! And of course Mike, who has put up with us and our random chatter, better than i ever imagined he would. It has been an absolute pleasure to work along side him, and i hope i get the chance to do it again.
Thank you all for reading 🙂