Evening,
Today we washed our plugs, as before in 1X wash buffer 4 times for an hour each. We then separated our 5 plugs into separate eppendorfs, and put 3 in storage in fresh 1X wash buffer. To the 2 remaining plugs we added 0.1X wash buffer for an hour.
To one we removed the wash buffer and added 1ml of Spe1 buffer to saturate for an hour. We then added fresh Spe1 buffer, Spe1 enzyme and BSA, and incubated overnight at 37C.
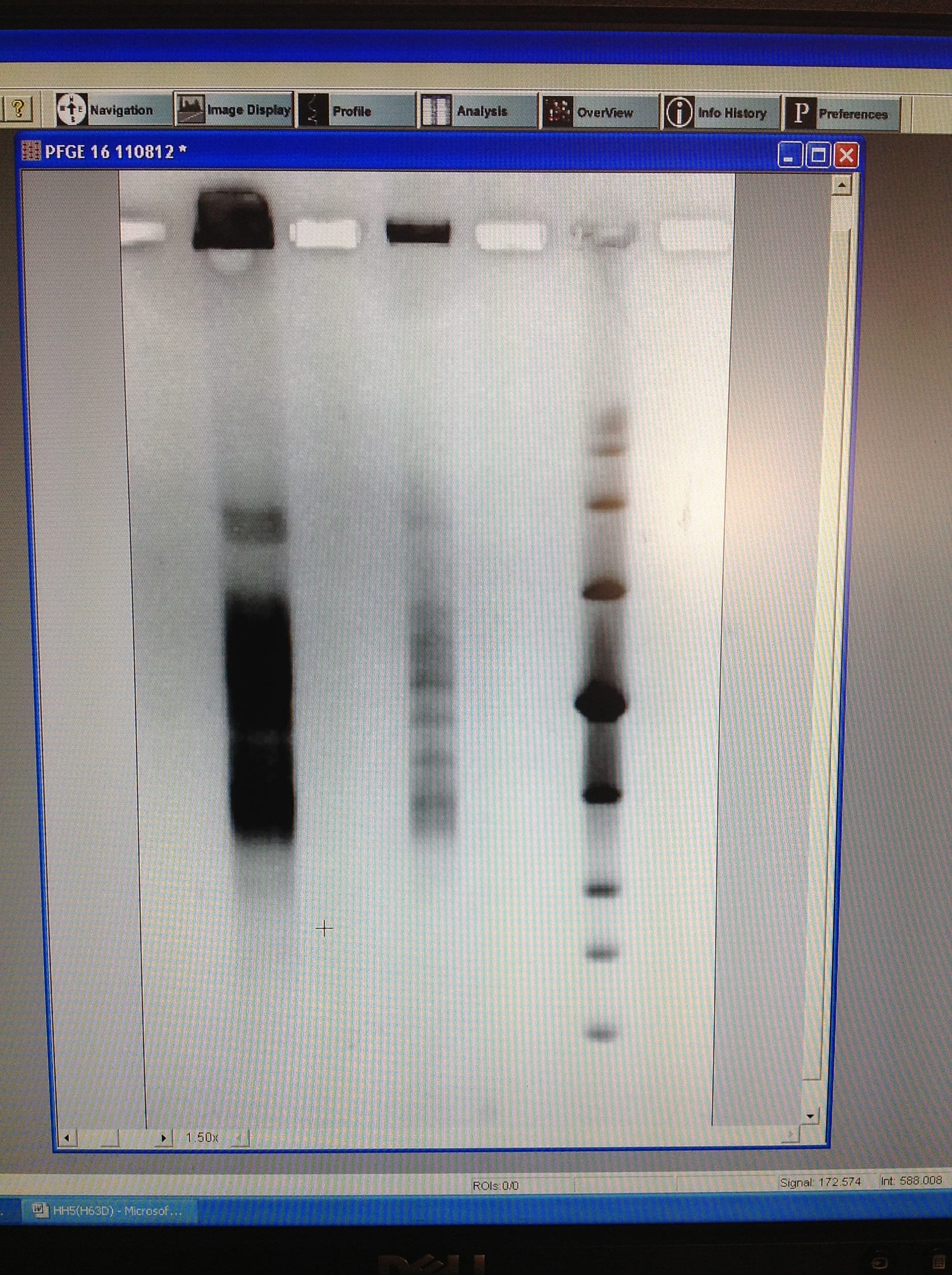
We set up a new gel in order to run the unrestriction treated plug, to ensure that the DNA within the samples are viable for use. If we see one large band just below the wells, that is as clear or clearer as our best previous gel run, then we will run a gel with the same settings, but with restriction enzyme treated plugs.
We made up 3L of TAE 0.5X and a 1% gel. We removed the wash buffer from the other plug and added 1ml of TAE 0.5X buffer to saturate it. Once the gel had set, we cut and placed 2 non restriction enzyme treated plug sections and 2 sections of the new low range PFG marker into the wells and sealed with agarose. We ran this gel at the low range PFG marker settings: 1% agarose gel, 15 hours, 1-12 second switch interval ramping and 6V/cm.
I also filmed with Sophia and Barney, regarding their experiences in the lab so far! 🙂
We will remove this gel tomorrow, stain it and view it. This will help us to decide whether to run the restriction enzyme plug, with the same low range PFG marker settings.
See you all tomorrow!